
Data quality control
Sequencing technologies all have some amount of error. Some of this error happens during PCR and some happens during sequencing. There are lots of ways to filter out errors including:
- Removing sequences/bases that the sequencer indicates are low quality. This information is typically in the .fastq files returned by the sequencer. There are many programs, such as trimmomatic, that can use the quality scores in these files to filter out low-quality sequences.
- Clustering similar sequences together. Many erroneous sequences are very similar to the true sequences, so they will be incorporated into the same OTU as the true sequence. In that sense, clustering into OTUs will hide some sequencing error.
- Removal of chimeric sequences. “Chimeras” are more significant errors that occur during PCR when an incomplete amplicon acts as a primer for a different template in a subsequent cycle. Most amplicon metagenomic pipelines (e.g. QIIME, mothur, usearch) have functions to detect and remove chimeras.
- Removing low-abundance sequences. Most of the time, erroneous sequences will occur much less often than the true sequence. Simply removing any unique sequence / OTU that appears less than some minimum number of times is an effective way to remove these errors. OTUs represented by only a single sequence are commonly called singletons. So when you hear things like “singletons were removed” and means all OTUs composed of only a single sequence were removed.
- Rarefying read counts. Usually, we try to make each sample in a sequencing run have the same number of reads sequenced, but that does not always happen. When a sample has many more reads than another sample, its apparent diversity can be artificially inflated since rare taxa are more likely to be found. To avoid this, the reads are subsampled to a fixed number or “rarefied”. This technique can throw out a lot of data, so it is not always appropriate (McMurdie and Holmes (2014)).
- Converting counts to presence/absence. Due to PCR biases and the nature of compositional data, many researchers advocate not using read count information at all. Instead, they recommend converting counts to simply “present” or “absent”.
Since we are only working with abundance matrices in these tutorials, the first three types of quality control have already been done (ideally), but we can still remove low-abundance OTUs and rarefy the counts of the remaining OTUs.
Load example data
If you are starting the workshop at this section, or had problems
running code in a previous section, use the following to load the data
used in this section. You can download the “filtered_data.Rdata” file
here.
If obj and sample_data are already in your
environment, you can ignore this and proceed.
load("filtered_data.Rdata")Removing low-abundance counts
The easiest way to get rid of some error in your data is to throw out
any count information below some threshold. The threshold is up to you;
removing singletons or
doubletons
is common, but lets be more conservative and remove any counts less than
10. The zero_low_counts counts can be used to convert any
counts less than some number to zero.
library(metacoder)
obj$data$otu_counts <- zero_low_counts(obj, "otu_counts", min_count = 10,
other_cols = TRUE) # keep OTU_ID column## No `cols` specified, so using all numeric columns:
## M1981P563, M1977P1709, M1980P502, M1981P606 ... M1955P747, M1978P342, M1955P778## Zeroing 576512 of 6331728 counts less than 10.print(obj)## <Taxmap>
## 704 taxa: aad. Bacteria, aaf. Proteobacteria ... cgz. S24-7, chs. Alcanivoracaceae
## 704 edges: NA->aad, aad->aaf, aad->aag, aad->aah ... amg->cgf, arh->cgz, apt->chs
## 1 data sets:
## otu_counts:
## # A tibble: 21,684 × 294
## taxon_id OTU_ID M1981P563 M1977P…¹ M1980…² M1981…³ M1981…⁴ M1981…⁵ M1981…⁶ M1982…⁷
## <chr> <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 ben 2 0 0 0 0 0 0 24 0
## 2 beo 3 10251 59 15857 309 134 90 11391 63
## 3 ben 4 1447 2496 2421 3331 143 1847 2120 2048
## # … with 21,681 more rows, 284 more variables: M1979P447 <dbl>, M1982P701 <dbl>,
## # M1982P661 <dbl>, M1978P293 <dbl>, M1980P462 <dbl>, M1555P221 <dbl>,
## # M1980P471 <dbl>, M1979P422 <dbl>, M1982P655 <dbl>, M1554P182 <dbl>, …, and
## # abbreviated variable names ¹M1977P1709, ²M1980P502, ³M1981P606, ⁴M1981P599,
## # ⁵M1981P611, ⁶M1981P587, ⁷M1982P729
## 0 functions:
That set all read counts less than 10 to zero. It did not filter out
any OTUs or their associated taxa however. We can do that using
filter_obs, which is used to filter data associated with a
taxonomy in a taxmap object. First lets find which OTUs now
have now reads associated with them.
no_reads <- rowSums(obj$data$otu_counts[, sample_data$SampleID]) == 0
sum(no_reads) # when `sum` is used on a TRUE/FALSE vector it counts TRUEs## [1] 14990
So now 14,990 of 21,684 OTUs have no reads. We can remove them and the taxa they are associated with like so:
obj <- filter_obs(obj, "otu_counts", ! no_reads, drop_taxa = TRUE)
print(obj)## <Taxmap>
## 489 taxa: aad. Bacteria, aaf. Proteobacteria ... ccz. Enterococcaceae, cfp. Dermacoccaceae
## 489 edges: NA->aad, aad->aaf, aad->aag, aad->aah ... amj->byk, aqy->ccz, amj->cfp
## 1 data sets:
## otu_counts:
## # A tibble: 6,694 × 294
## taxon_id OTU_ID M1981P563 M1977P…¹ M1980…² M1981…³ M1981…⁴ M1981…⁵ M1981…⁶ M1982…⁷
## <chr> <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 ben 2 0 0 0 0 0 0 24 0
## 2 beo 3 10251 59 15857 309 134 90 11391 63
## 3 ben 4 1447 2496 2421 3331 143 1847 2120 2048
## # … with 6,691 more rows, 284 more variables: M1979P447 <dbl>, M1982P701 <dbl>,
## # M1982P661 <dbl>, M1978P293 <dbl>, M1980P462 <dbl>, M1555P221 <dbl>,
## # M1980P471 <dbl>, M1979P422 <dbl>, M1982P655 <dbl>, M1554P182 <dbl>, …, and
## # abbreviated variable names ¹M1977P1709, ²M1980P502, ³M1981P606, ⁴M1981P599,
## # ⁵M1981P611, ⁶M1981P587, ⁷M1982P729
## 0 functions:
Rarefaction
Rarefaction is used to simulate even numbers of reads per sample. Even sampling is important for at least two reasons:
- When comparing diversity of samples, more samples make it more likely to observe rare species. This will have a larger effect on some diversity indexes than others, depending on how they weigh rare species.
- When comparing the similarity of samples, the presence of rare species due to higher sampling depth in one sample but not another can make the two samples appear more different than they actually are.
Therefore, when comparing the diversity or similarity of samples, it is important to take into account differences in sampling depth.
Rarefying can waste a lot of data and is not needed in many cases. See McMurdie and Holmes (2014) for an in-depth discussion on alternatives to rarefaction. We cover it here because it is a popular technique.
Typically, the rarefaction depth chosen is the minimum sample depth. If the minimum depth is very small, the samples with the smallest depth can be removed and the minimum depth of the remaining samples can be used.
Lets take a look at the distribution of read depths of our samples:
hist(colSums(obj$data$otu_counts[, sample_data$SampleID]))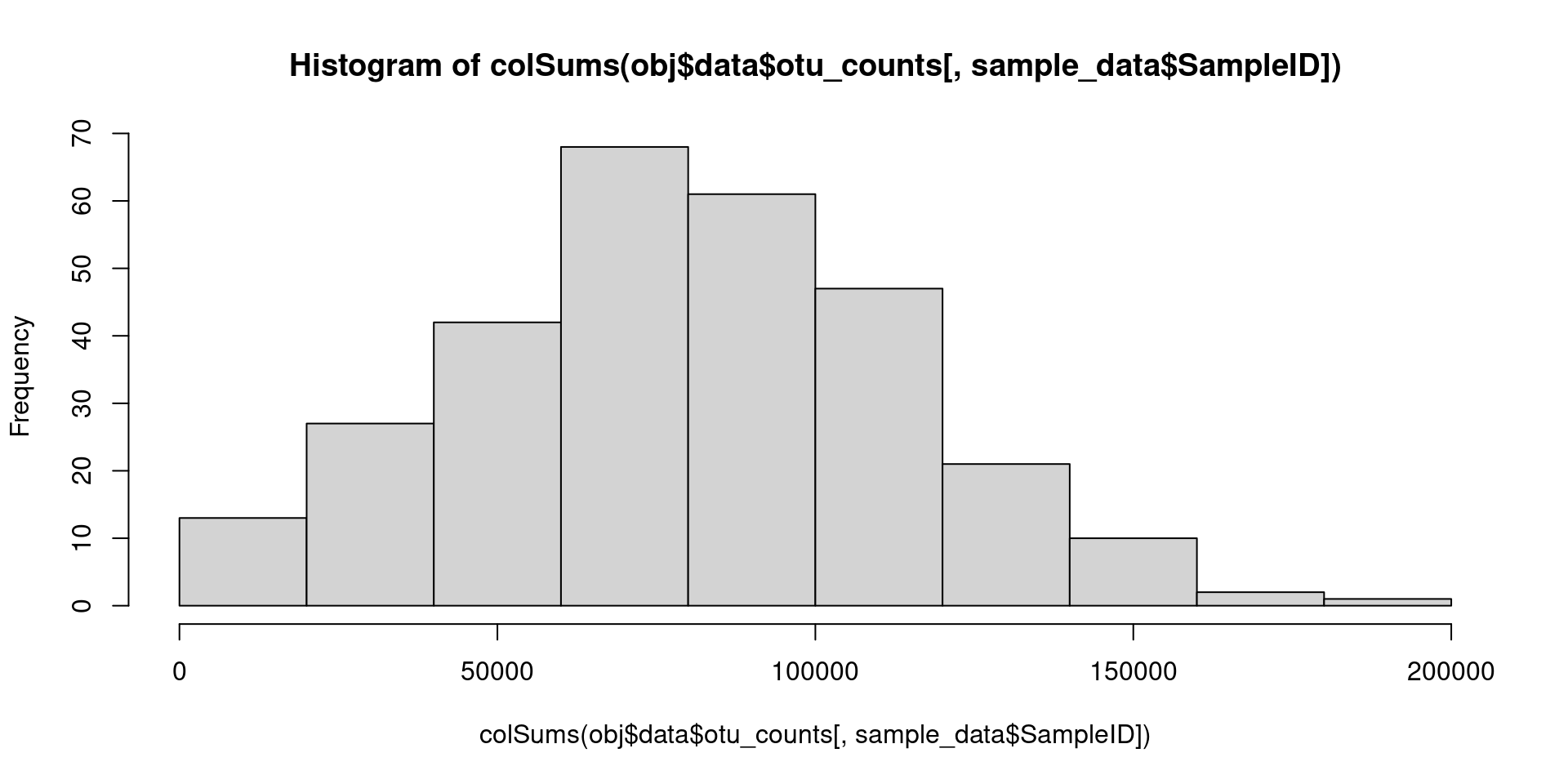
We have a minimum depth of 1857, a median of 78767 and a maximum
depth of 1.89353^{5}. We could try to remove one or two of the samples
with the smallest depth, since it seems like a waste to throw out so
much data, but for this tutorial we will just rarefy to the minimum of
1857 reads. The vegan package implements many functions to
help with rarefying data, but we will use a function from
metacoder that calls the functions from vegan
and re-formats the input and outputs to make them easier to use with the
way our data is formatted.
obj$data$otu_rarefied <- rarefy_obs(obj, "otu_counts", other_cols = TRUE)## No `cols` specified, so using all numeric columns:
## M1981P563, M1977P1709, M1980P502, M1981P606 ... M1955P747, M1978P342, M1955P778## Rarefying to 1857 since that is the lowest sample total.print(obj)## <Taxmap>
## 489 taxa: aad. Bacteria, aaf. Proteobacteria ... ccz. Enterococcaceae, cfp. Dermacoccaceae
## 489 edges: NA->aad, aad->aaf, aad->aag, aad->aah ... amj->byk, aqy->ccz, amj->cfp
## 2 data sets:
## otu_counts:
## # A tibble: 6,694 × 294
## taxon_id OTU_ID M1981P563 M1977P…¹ M1980…² M1981…³ M1981…⁴ M1981…⁵ M1981…⁶ M1982…⁷
## <chr> <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 ben 2 0 0 0 0 0 0 24 0
## 2 beo 3 10251 59 15857 309 134 90 11391 63
## 3 ben 4 1447 2496 2421 3331 143 1847 2120 2048
## # … with 6,691 more rows, 284 more variables: M1979P447 <dbl>, M1982P701 <dbl>,
## # M1982P661 <dbl>, M1978P293 <dbl>, M1980P462 <dbl>, M1555P221 <dbl>,
## # M1980P471 <dbl>, M1979P422 <dbl>, M1982P655 <dbl>, M1554P182 <dbl>, …, and
## # abbreviated variable names ¹M1977P1709, ²M1980P502, ³M1981P606, ⁴M1981P599,
## # ⁵M1981P611, ⁶M1981P587, ⁷M1982P729
## otu_rarefied:
## # A tibble: 6,694 × 294
## taxon_id OTU_ID M1981P563 M1977P…¹ M1980…² M1981…³ M1981…⁴ M1981…⁵ M1981…⁶ M1982…⁷
## <chr> <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 ben 2 0 0 0 0 0 0 1 0
## 2 beo 3 402 4 428 5 134 0 347 2
## 3 ben 4 63 46 67 77 143 50 70 44
## # … with 6,691 more rows, 284 more variables: M1979P447 <dbl>, M1982P701 <dbl>,
## # M1982P661 <dbl>, M1978P293 <dbl>, M1980P462 <dbl>, M1555P221 <dbl>,
## # M1980P471 <dbl>, M1979P422 <dbl>, M1982P655 <dbl>, M1554P182 <dbl>, …, and
## # abbreviated variable names ¹M1977P1709, ²M1980P502, ³M1981P606, ⁴M1981P599,
## # ⁵M1981P611, ⁶M1981P587, ⁷M1982P729
## 0 functions:
This probably means that some OTUs now have no reads in the rarefied
dataset. Lets remove those like we did before. However, since there is
now two tables, we should not remove any taxa since they still might be
used in the rarefied table, so we will not add the
drop_taxa = TRUE option this time.
no_reads <- rowSums(obj$data$otu_rarefied[, sample_data$SampleID]) == 0
obj <- filter_obs(obj, "otu_rarefied", ! no_reads)
print(obj)## <Taxmap>
## 489 taxa: aad. Bacteria, aaf. Proteobacteria ... ccz. Enterococcaceae, cfp. Dermacoccaceae
## 489 edges: NA->aad, aad->aaf, aad->aag, aad->aah ... amj->byk, aqy->ccz, amj->cfp
## 2 data sets:
## otu_counts:
## # A tibble: 6,694 × 294
## taxon_id OTU_ID M1981P563 M1977P…¹ M1980…² M1981…³ M1981…⁴ M1981…⁵ M1981…⁶ M1982…⁷
## <chr> <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 ben 2 0 0 0 0 0 0 24 0
## 2 beo 3 10251 59 15857 309 134 90 11391 63
## 3 ben 4 1447 2496 2421 3331 143 1847 2120 2048
## # … with 6,691 more rows, 284 more variables: M1979P447 <dbl>, M1982P701 <dbl>,
## # M1982P661 <dbl>, M1978P293 <dbl>, M1980P462 <dbl>, M1555P221 <dbl>,
## # M1980P471 <dbl>, M1979P422 <dbl>, M1982P655 <dbl>, M1554P182 <dbl>, …, and
## # abbreviated variable names ¹M1977P1709, ²M1980P502, ³M1981P606, ⁴M1981P599,
## # ⁵M1981P611, ⁶M1981P587, ⁷M1982P729
## otu_rarefied:
## # A tibble: 5,025 × 294
## taxon_id OTU_ID M1981P563 M1977P…¹ M1980…² M1981…³ M1981…⁴ M1981…⁵ M1981…⁶ M1982…⁷
## <chr> <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 ben 2 0 0 0 0 0 0 1 0
## 2 beo 3 402 4 428 5 134 0 347 2
## 3 ben 4 63 46 67 77 143 50 70 44
## # … with 5,022 more rows, 284 more variables: M1979P447 <dbl>, M1982P701 <dbl>,
## # M1982P661 <dbl>, M1978P293 <dbl>, M1980P462 <dbl>, M1555P221 <dbl>,
## # M1980P471 <dbl>, M1979P422 <dbl>, M1982P655 <dbl>, M1554P182 <dbl>, …, and
## # abbreviated variable names ¹M1977P1709, ²M1980P502, ³M1981P606, ⁴M1981P599,
## # ⁵M1981P611, ⁶M1981P587, ⁷M1982P729
## 0 functions:
Proportions
An easy alternative to rarefaction is to convert read counts to proportions of reads per-sample. This makes samples directly comparable, but does not solve the problem with inflated diversity estimates discussed in the rarefaction section. See McMurdie and Holmes (2014) for an in-depth discussion on the relative merits of proportions, rarefaction, and other (better) methods.
To convert counts to proportions in metacoder, we can
use the calc_obs_props function:
obj$data$otu_props <- calc_obs_props(obj, "otu_counts", other_cols = TRUE)## No `cols` specified, so using all numeric columns:
## M1981P563, M1977P1709, M1980P502, M1981P606 ... M1955P747, M1978P342, M1955P778## Calculating proportions from counts for 292 columns for 6694 observations.print(obj)## <Taxmap>
## 489 taxa: aad. Bacteria, aaf. Proteobacteria ... ccz. Enterococcaceae, cfp. Dermacoccaceae
## 489 edges: NA->aad, aad->aaf, aad->aag, aad->aah ... amj->byk, aqy->ccz, amj->cfp
## 3 data sets:
## otu_counts:
## # A tibble: 6,694 × 294
## taxon_id OTU_ID M1981P563 M1977P…¹ M1980…² M1981…³ M1981…⁴ M1981…⁵ M1981…⁶ M1982…⁷
## <chr> <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 ben 2 0 0 0 0 0 0 24 0
## 2 beo 3 10251 59 15857 309 134 90 11391 63
## 3 ben 4 1447 2496 2421 3331 143 1847 2120 2048
## # … with 6,691 more rows, 284 more variables: M1979P447 <dbl>, M1982P701 <dbl>,
## # M1982P661 <dbl>, M1978P293 <dbl>, M1980P462 <dbl>, M1555P221 <dbl>,
## # M1980P471 <dbl>, M1979P422 <dbl>, M1982P655 <dbl>, M1554P182 <dbl>, …, and
## # abbreviated variable names ¹M1977P1709, ²M1980P502, ³M1981P606, ⁴M1981P599,
## # ⁵M1981P611, ⁶M1981P587, ⁷M1982P729
## otu_rarefied:
## # A tibble: 5,025 × 294
## taxon_id OTU_ID M1981P563 M1977P…¹ M1980…² M1981…³ M1981…⁴ M1981…⁵ M1981…⁶ M1982…⁷
## <chr> <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 ben 2 0 0 0 0 0 0 1 0
## 2 beo 3 402 4 428 5 134 0 347 2
## 3 ben 4 63 46 67 77 143 50 70 44
## # … with 5,022 more rows, 284 more variables: M1979P447 <dbl>, M1982P701 <dbl>,
## # M1982P661 <dbl>, M1978P293 <dbl>, M1980P462 <dbl>, M1555P221 <dbl>,
## # M1980P471 <dbl>, M1979P422 <dbl>, M1982P655 <dbl>, M1554P182 <dbl>, …, and
## # abbreviated variable names ¹M1977P1709, ²M1980P502, ³M1981P606, ⁴M1981P599,
## # ⁵M1981P611, ⁶M1981P587, ⁷M1982P729
## otu_props:
## # A tibble: 6,694 × 294
## taxon_id OTU_ID M1981P563 M1977P…¹ M1980…² M1981…³ M1981…⁴ M1981…⁵ M1981…⁶ M1982…⁷
## <chr> <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 ben 2 0 0 0 0 0 0 4.09e-4 0
## 2 beo 3 0.200 0.000448 0.237 0.00327 0.0722 0.00101 1.94e-1 6.68e-4
## 3 ben 4 0.0282 0.0189 0.0362 0.0352 0.0770 0.0207 3.61e-2 2.17e-2
## # … with 6,691 more rows, 284 more variables: M1979P447 <dbl>, M1982P701 <dbl>,
## # M1982P661 <dbl>, M1978P293 <dbl>, M1980P462 <dbl>, M1555P221 <dbl>,
## # M1980P471 <dbl>, M1979P422 <dbl>, M1982P655 <dbl>, M1554P182 <dbl>, …, and
## # abbreviated variable names ¹M1977P1709, ²M1980P502, ³M1981P606, ⁴M1981P599,
## # ⁵M1981P611, ⁶M1981P587, ⁷M1982P729
## 0 functions:
Rarefaction curves
The relationship between the number of reads and the number of OTUs
can be described using
rarefaction
curves. This is actually different concept than rarefaction, and
it is done for different reasons. Rarefaction is a subsampling technique
meant to correct for uneven sample size whereas rarefaction curves are
used to estimate whether all the diveristy of the true community was
captured.
Each line represents a different sample (i.e. column) and shows how many
OTUs are found in a random subsets of different numbers of reads. The
rarecurve function from the vegan package will
do the random sub-sampling and plot the results for an abundance matrix.
For example, the code below plots the rarefaction curve for a single
sample:
library(vegan)## Loading required package: permute## Loading required package: lattice## This is vegan 2.6-2rarecurve(t(obj$data$otu_counts[, "M1981P563"]), step = 20,
sample = min(colSums(obj$data$otu_counts[, sample_data$SampleID])),
col = "blue", cex = 1.5)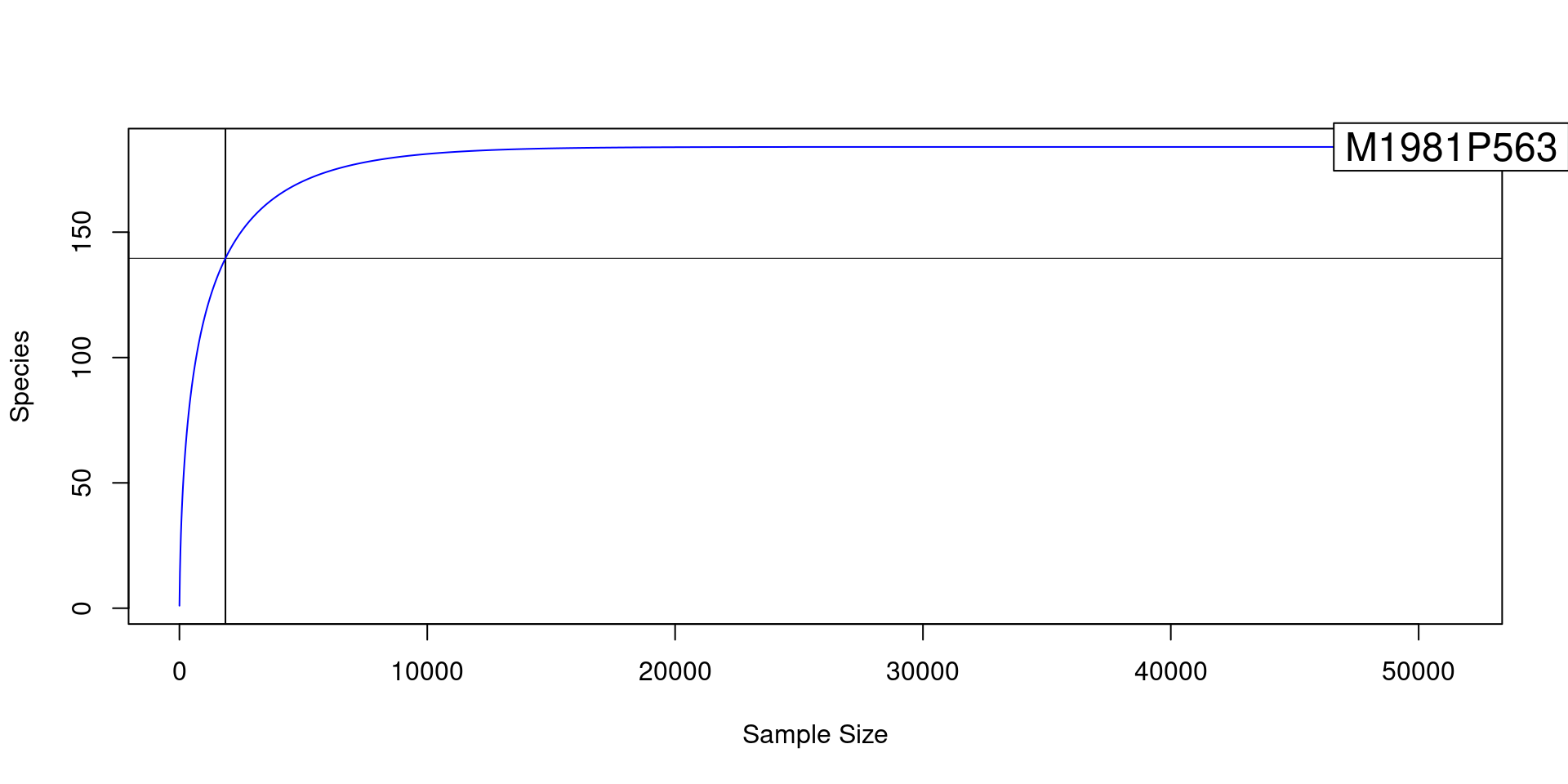
For this sample, few new OTUs are observed after about 20,000 reads.
Since the number of OTUs found flattens out, it suggests that the sample
had sufficient reads to capture most of the diversity (or reach a point
of diminishing returns). Like other functions in vegan,
rarecurve expects samples to in rows instead of columns, so
we had to
transpose
(make rows columns) the matrix with the t function before
passing it to rarecurve. If you plot all the samples, which
takes a few minutes, it looks like this:
Converting to presence/absence
Many researchers believe that read depth is not informative due to PCR and sequencing biases. Therefore, instead of comparing read counts, the counts can be converted to presence or absence of an OTU in a given sample. This can be done like so:
counts_to_presence(obj, "otu_rarefied")## No `cols` specified, so using all numeric columns:
## M1981P563, M1977P1709, M1980P502, M1981P606 ... M1955P747, M1978P342, M1955P778## # A tibble: 5,025 × 293
## taxon_id M1981P…¹ M1977…² M1980…³ M1981…⁴ M1981…⁵ M1981…⁶ M1981…⁷ M1982…⁸ M1979…⁹ M1982…˟ M1982…˟
## <chr> <lgl> <lgl> <lgl> <lgl> <lgl> <lgl> <lgl> <lgl> <lgl> <lgl> <lgl>
## 1 ben FALSE FALSE FALSE FALSE FALSE FALSE TRUE FALSE FALSE FALSE TRUE
## 2 beo TRUE TRUE TRUE TRUE TRUE FALSE TRUE TRUE TRUE TRUE TRUE
## 3 ben TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE
## 4 bep TRUE TRUE TRUE TRUE FALSE TRUE TRUE TRUE TRUE TRUE FALSE
## 5 beq TRUE TRUE TRUE TRUE FALSE TRUE TRUE TRUE TRUE TRUE FALSE
## 6 amk TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE
## 7 bes FALSE TRUE FALSE TRUE FALSE TRUE TRUE TRUE TRUE TRUE FALSE
## 8 bet TRUE TRUE TRUE TRUE TRUE FALSE TRUE FALSE TRUE TRUE TRUE
## 9 beu TRUE TRUE TRUE TRUE FALSE FALSE TRUE FALSE TRUE TRUE FALSE
## 10 bev FALSE TRUE FALSE TRUE FALSE TRUE TRUE TRUE TRUE TRUE FALSE
## # … with 5,015 more rows, 281 more variables: M1978P293 <lgl>, M1980P462 <lgl>, M1555P221 <lgl>,
## # M1980P471 <lgl>, M1979P422 <lgl>, M1982P655 <lgl>, M1554P182 <lgl>, M1981P623 <lgl>,
## # M1555P239 <lgl>, M1958P1020 <lgl>, …, and abbreviated variable names ¹M1981P563, ²M1977P1709,
## # ³M1980P502, ⁴M1981P606, ⁵M1981P599, ⁶M1981P611, ⁷M1981P587, ⁸M1982P729, ⁹M1979P447, ˟M1982P701,
## # ˟M1982P661
For this workshop however, we will use the read counts.
Exercises
In these exercises, we will be using the obj from the
analysis above. If you did not run the code above or had problems, run
the following code to get the objects used. You can download the
“clean_data.Rdata” file
here.
load("clean_data.Rdata")1a) Consider the following rarefaction curves:
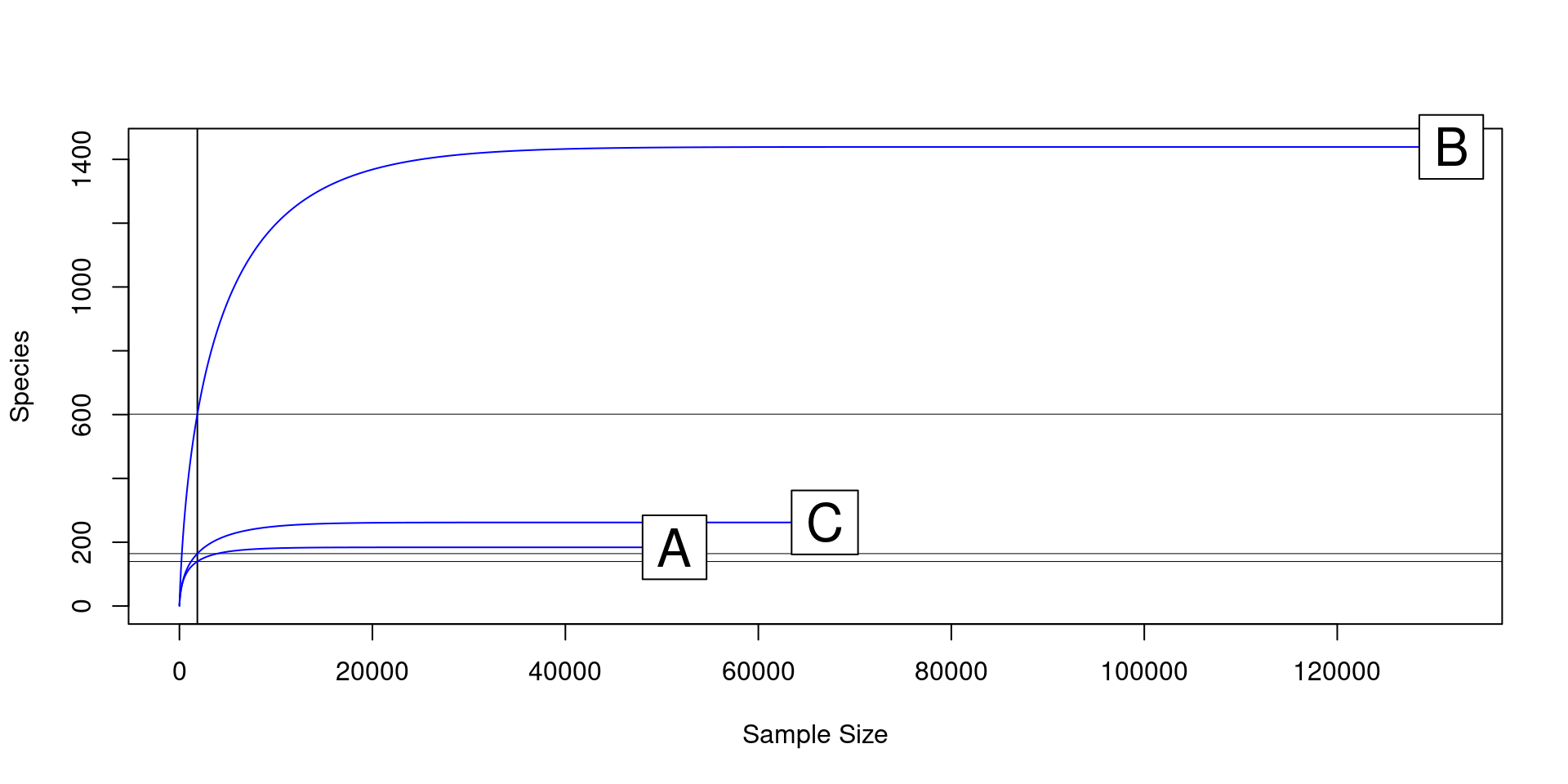
1b) Which sample is more diverse?
1c) Which sample needs the most reads to capture all of its diversity?
1d) What would be a sufficient read count to capture all of the diversity in these three samples?
2) Look at the documentation for
rarefy_obs. Rarefy the sample OTU counts to 1000 reads.
3) What are two ways of accounting for uneven sample depth?
4) What are some reasons that read counts might not correspond to species abundance in the original sample? Try to think of at least 3. There are 7 listed in the answer below and there are probably other reasons not listed.