Assign taxonomy functions
Usage
assign_tax(
analysis_setup,
asv_abund_matrix,
retrieve_files = FALSE,
overwrite_existing = FALSE,
db_rps10 = "oomycetedb.fasta",
db_its = "fungidb.fasta",
db_16S = "bacteriadb.fasta",
db_other1 = "otherdb1.fasta",
db_other2 = "otherdb2.fasta"
)Arguments
- analysis_setup
An object containing directory paths and data tables, produced by the
prepare_readsfunction- asv_abund_matrix
The final abundance matrix containing amplified sequence variants
- retrieve_files
Logical, TRUE/FALSE whether to copy files from the temp directory to the output directory. Default is FALSE.
- overwrite_existing
Logical, indicating whether to remove or overwrite existing files and directories from previous runs. Default is
FALSE.- db_rps10
The reference database for the rps10 metabarcode
- db_its
The reference database for the ITS metabarcode
- db_16S
The SILVA 16S-rRNA reference database provided by the user
- db_other1
The reference database for other metabarcode 1 (assumes format is like SILVA DB entries)
- db_other2
The reference database for other metabarcode 2 (assumes format is like SILVA DB entries)
Details
At this point, 'DADA2' function assignTaxonomy is used to assign taxonomy to the inferred ASVs.
Examples
# \donttest{
# Assign taxonomies to ASVs on by metabarcode
analysis_setup <- prepare_reads(
data_directory = system.file("extdata", package = "demulticoder"),
output_directory = tempdir(),
overwrite_existing = TRUE
)
#> Existing files found in the output directory. Overwriting existing files.
#> Rows: 2 Columns: 25
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: ","
#> chr (3): primer_name, forward, reverse
#> dbl (18): minCutadaptlength, maxN, maxEE_forward, maxEE_reverse, truncLen_fo...
#> lgl (4): already_trimmed, count_all_samples, multithread, verbose
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Rows: 2 Columns: 25
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: ","
#> chr (3): primer_name, forward, reverse
#> dbl (18): minCutadaptlength, maxN, maxEE_forward, maxEE_reverse, truncLen_fo...
#> lgl (4): already_trimmed, count_all_samples, multithread, verbose
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Rows: 4 Columns: 3
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: ","
#> chr (3): sample_name, primer_name, organism
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Creating output directory: /tmp/RtmpAtZc28/demulticoder_run/prefiltered_sequences
 cut_trim(
analysis_setup,
cutadapt_path="/usr/bin/cutadapt",
overwrite_existing = TRUE
)
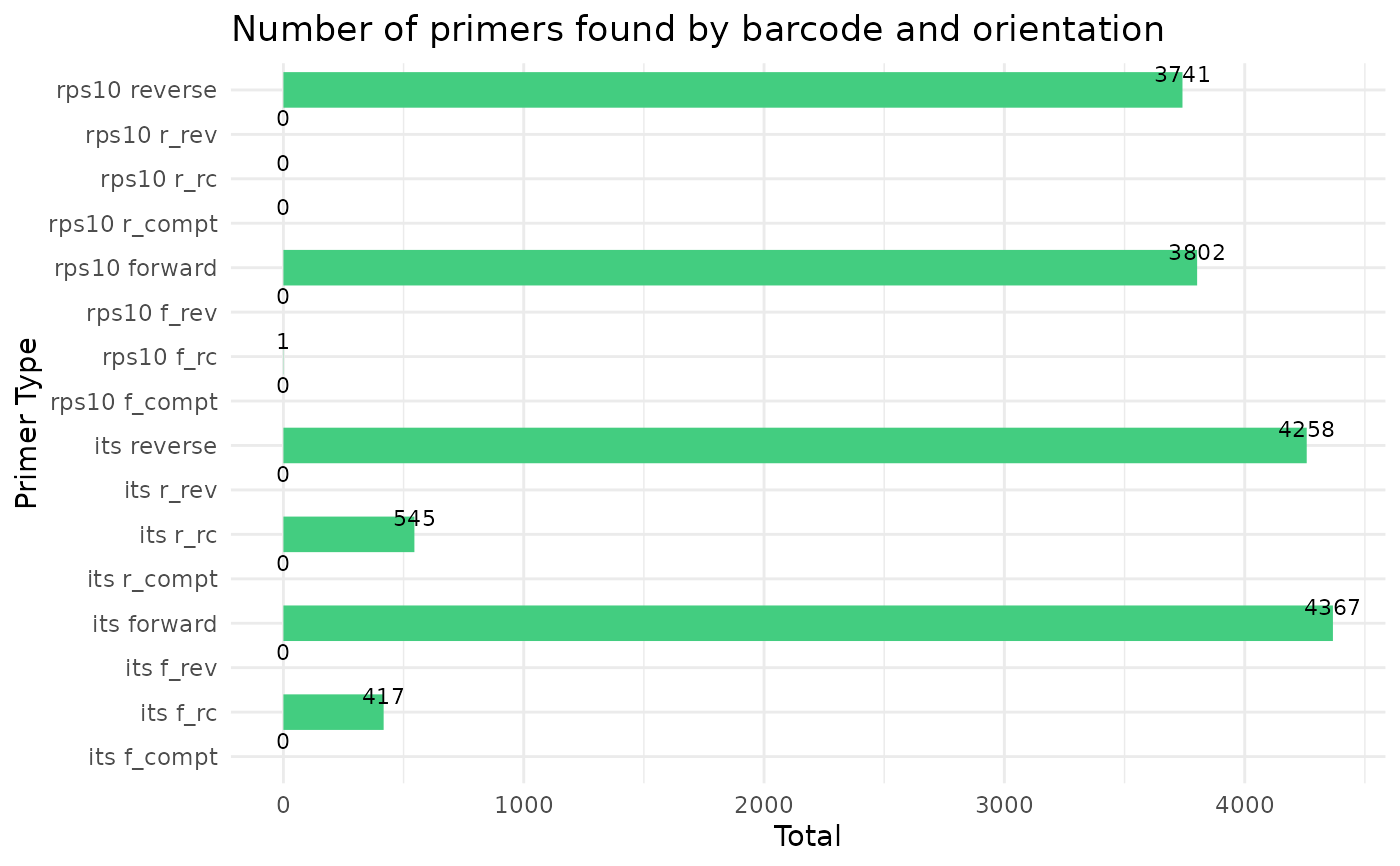
#> Running cutadapt 3.5 for its sequence data
cut_trim(
analysis_setup,
cutadapt_path="/usr/bin/cutadapt",
overwrite_existing = TRUE
)
#> Running cutadapt 3.5 for its sequence data
 #> Running cutadapt 3.5 for rps10 sequence data
#> Running cutadapt 3.5 for rps10 sequence data
 make_asv_abund_matrix(
analysis_setup,
overwrite_existing = TRUE
)
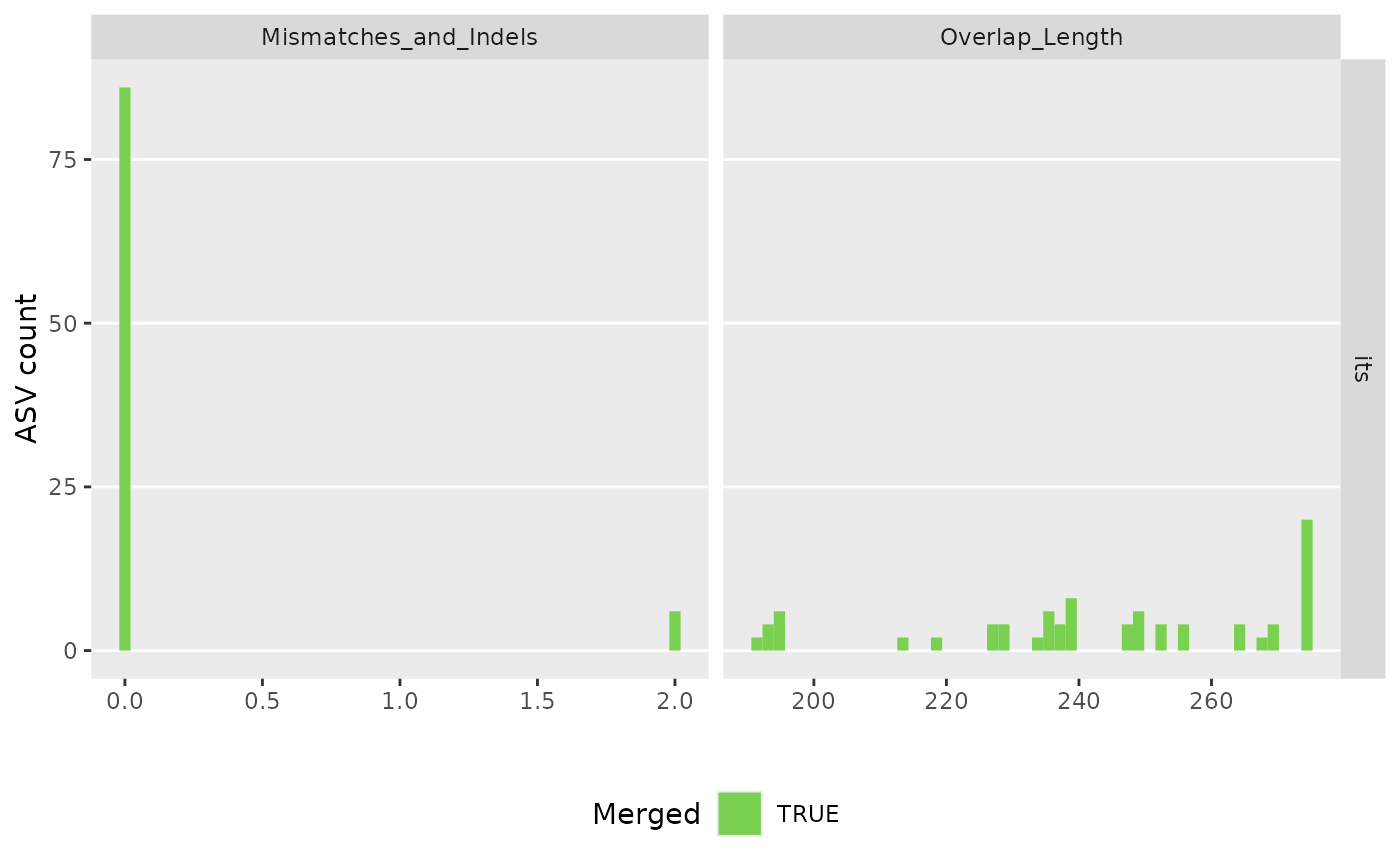
#> 80608 total bases in 307 reads from 2 samples will be used for learning the error rates.
#> Error rate plot for the Forward read of primer pair its
#> Warning: log-10 transformation introduced infinite values.
#> Sample 1 - 163 reads in 84 unique sequences.
#> Sample 2 - 144 reads in 96 unique sequences.
#> 82114 total bases in 307 reads from 2 samples will be used for learning the error rates.
#> Error rate plot for the Reverse read of primer pair its
#> Warning: log-10 transformation introduced infinite values.
#> Sample 1 - 163 reads in 128 unique sequences.
#> Sample 2 - 144 reads in 119 unique sequences.
make_asv_abund_matrix(
analysis_setup,
overwrite_existing = TRUE
)
#> 80608 total bases in 307 reads from 2 samples will be used for learning the error rates.
#> Error rate plot for the Forward read of primer pair its
#> Warning: log-10 transformation introduced infinite values.
#> Sample 1 - 163 reads in 84 unique sequences.
#> Sample 2 - 144 reads in 96 unique sequences.
#> 82114 total bases in 307 reads from 2 samples will be used for learning the error rates.
#> Error rate plot for the Reverse read of primer pair its
#> Warning: log-10 transformation introduced infinite values.
#> Sample 1 - 163 reads in 128 unique sequences.
#> Sample 2 - 144 reads in 119 unique sequences.
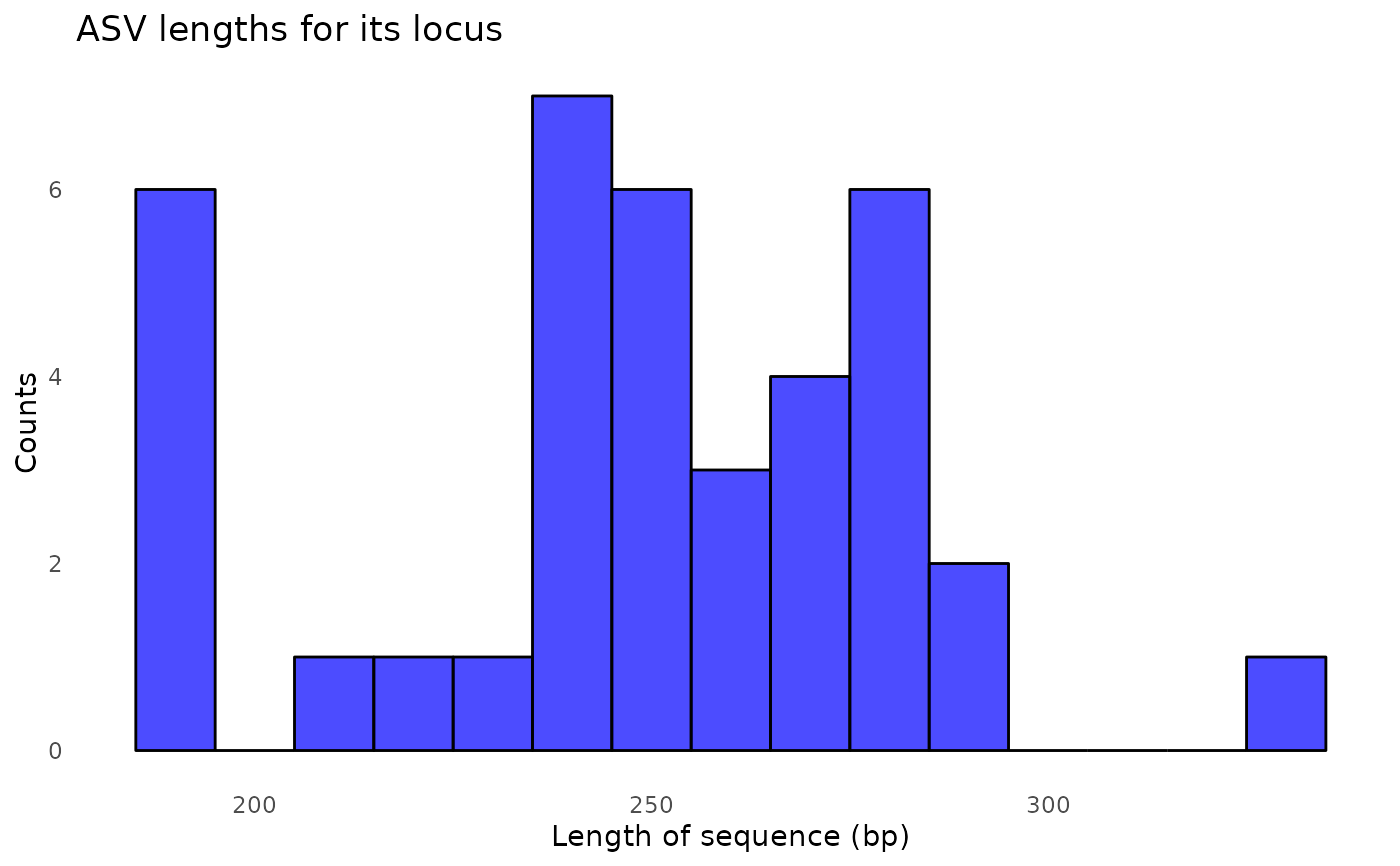
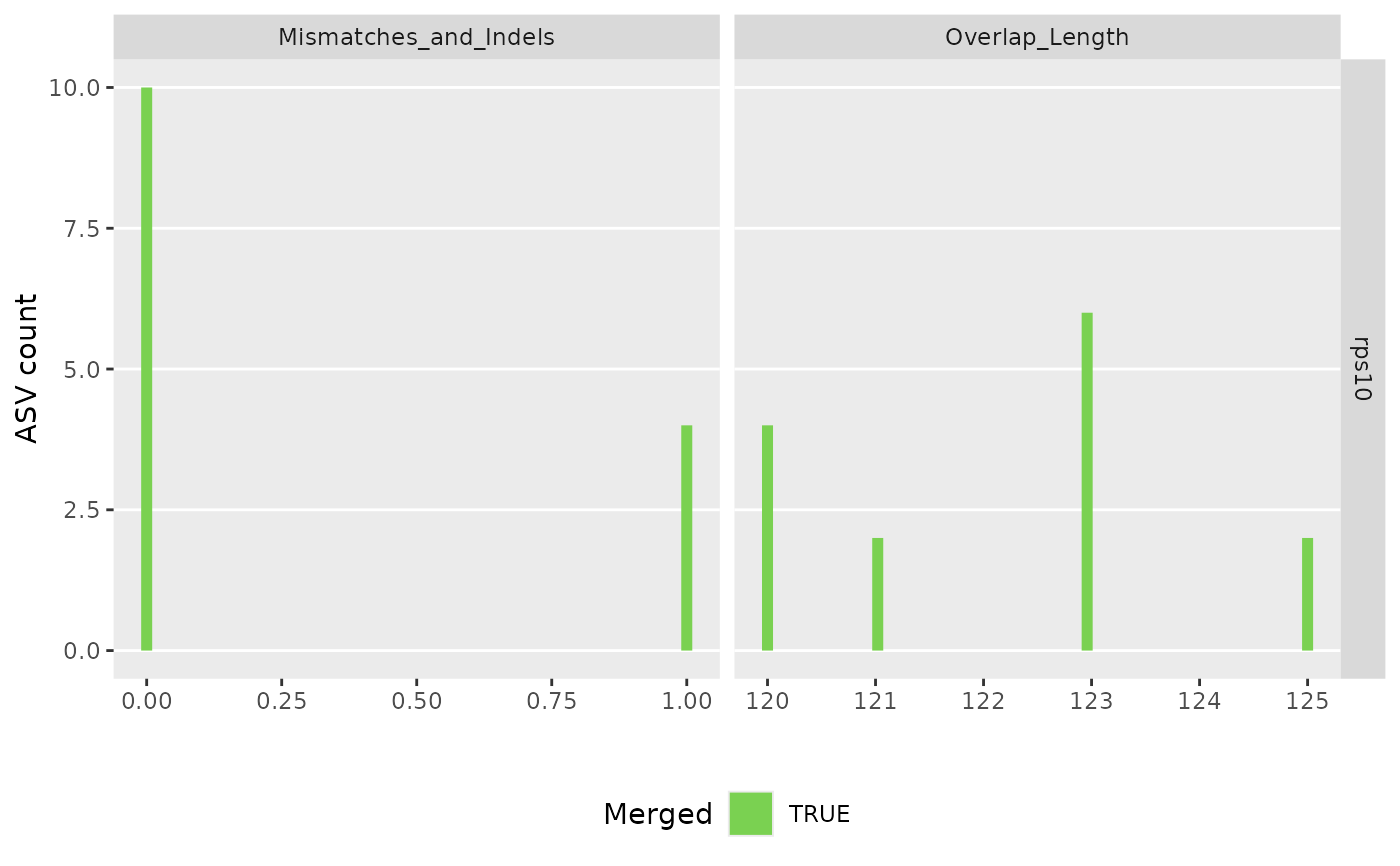 #> 91897 total bases in 327 reads from 2 samples will be used for learning the error rates.
#> Error rate plot for the Forward read of primer pair rps10
#> Warning: log-10 transformation introduced infinite values.
#> Sample 1 - 145 reads in 107 unique sequences.
#> Sample 2 - 182 reads in 133 unique sequences.
#> 91567 total bases in 327 reads from 2 samples will be used for learning the error rates.
#> Error rate plot for the Reverse read of primer pair rps10
#> Warning: log-10 transformation introduced infinite values.
#> Sample 1 - 145 reads in 114 unique sequences.
#> Sample 2 - 182 reads in 170 unique sequences.
#> 91897 total bases in 327 reads from 2 samples will be used for learning the error rates.
#> Error rate plot for the Forward read of primer pair rps10
#> Warning: log-10 transformation introduced infinite values.
#> Sample 1 - 145 reads in 107 unique sequences.
#> Sample 2 - 182 reads in 133 unique sequences.
#> 91567 total bases in 327 reads from 2 samples will be used for learning the error rates.
#> Error rate plot for the Reverse read of primer pair rps10
#> Warning: log-10 transformation introduced infinite values.
#> Sample 1 - 145 reads in 114 unique sequences.
#> Sample 2 - 182 reads in 170 unique sequences.
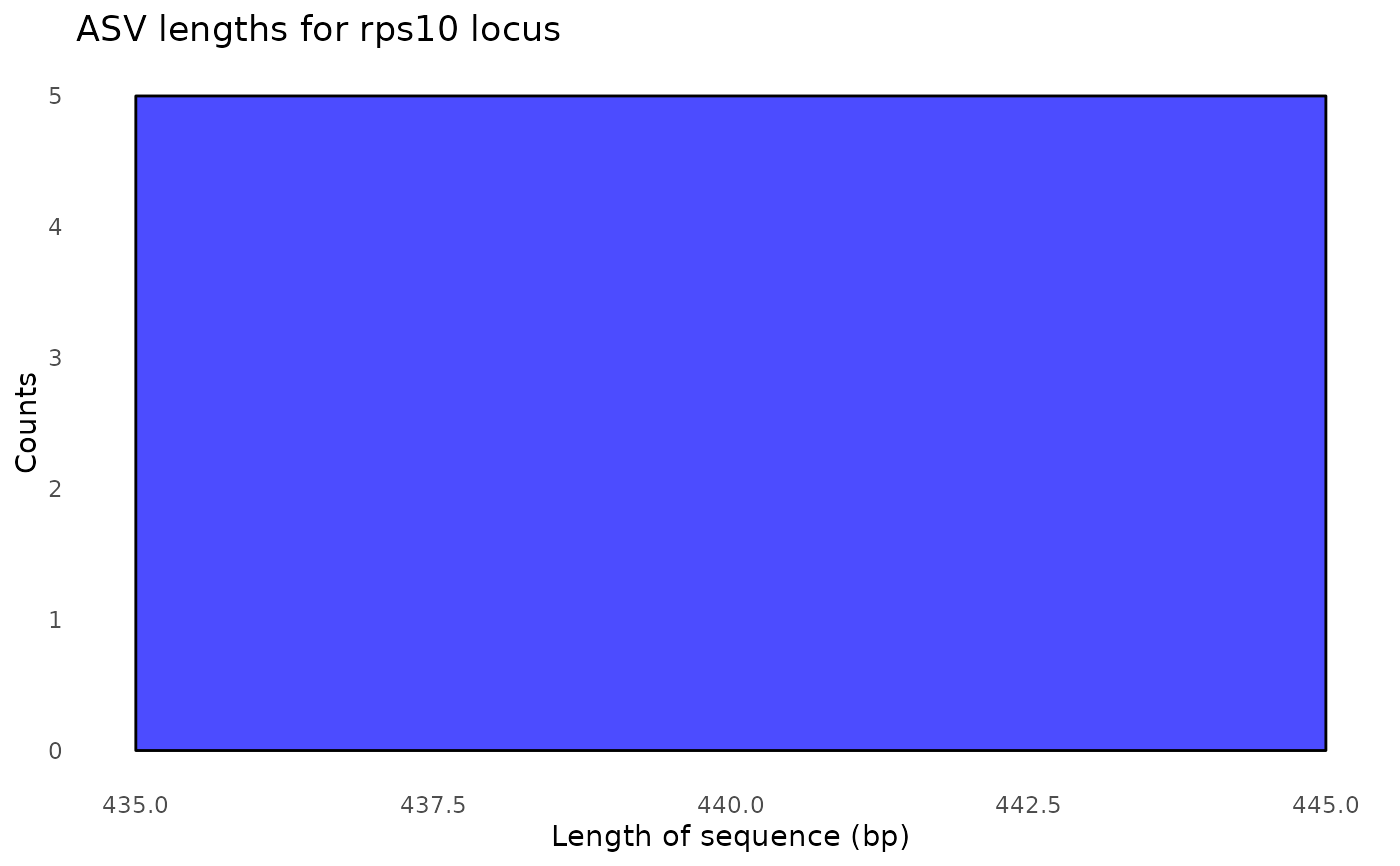
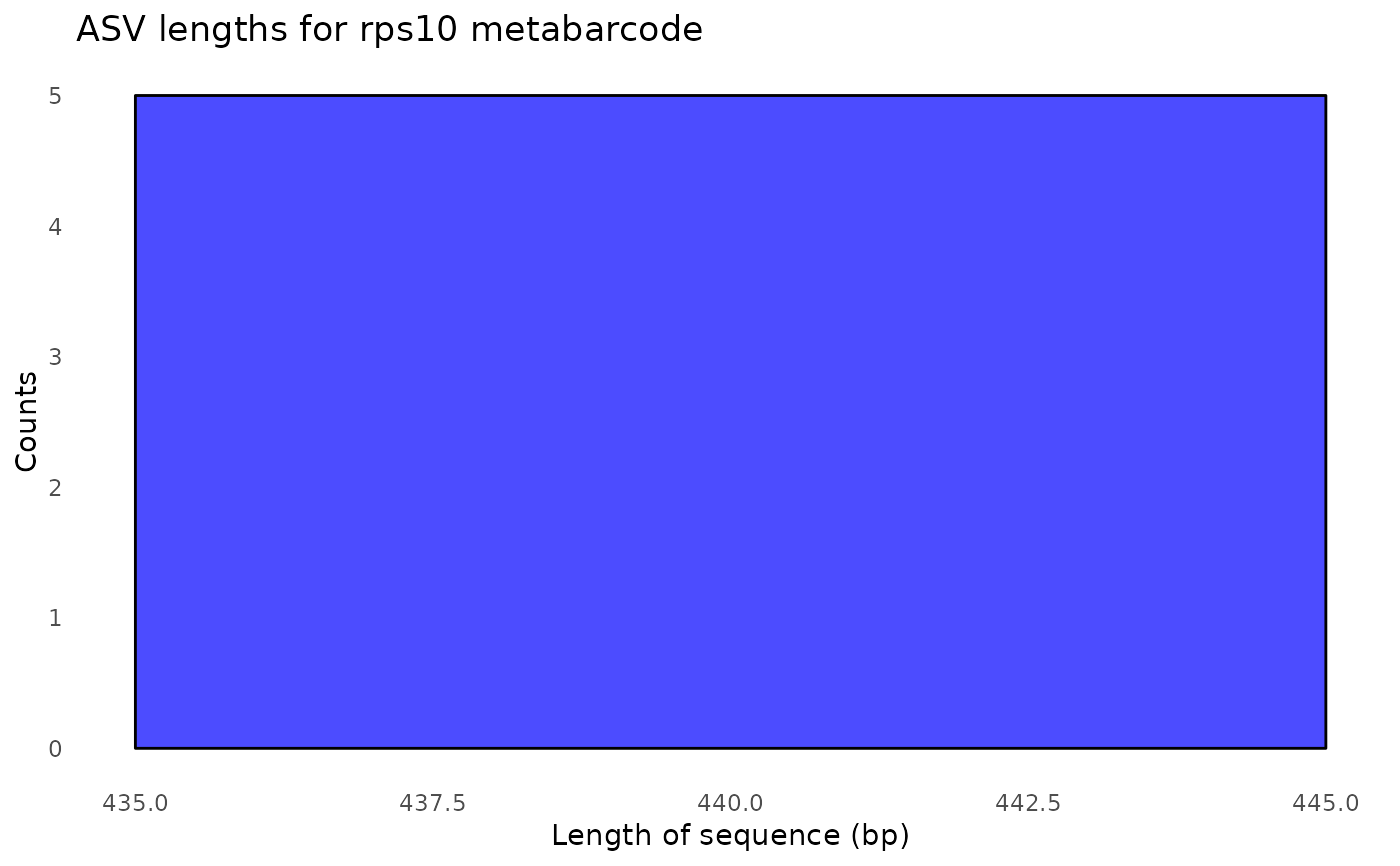 #> $its
#> [1] "/tmp/RtmpAtZc28/demulticoder_run/asvabund_matrixDADA2_its.RData"
#>
#> $rps10
#> [1] "/tmp/RtmpAtZc28/demulticoder_run/asvabund_matrixDADA2_rps10.RData"
#>
assign_tax(
analysis_setup,
asv_abund_matrix,
retrieve_files=FALSE,
overwrite_existing = TRUE
)
#> Tracking read counts:
#> samplename_barcode input filtered denoisedF denoisedR merged nonchim
#> 1 S1_its 299 163 146 141 132 132
#> 2 S2_its 235 144 113 99 99 99
#> Tracking read counts:
#> samplename_barcode input filtered denoisedF denoisedR merged nonchim
#> 1 S1_rps10 196 145 145 145 145 145
#> 2 S2_rps10 253 182 181 181 181 181
# }
#> $its
#> [1] "/tmp/RtmpAtZc28/demulticoder_run/asvabund_matrixDADA2_its.RData"
#>
#> $rps10
#> [1] "/tmp/RtmpAtZc28/demulticoder_run/asvabund_matrixDADA2_rps10.RData"
#>
assign_tax(
analysis_setup,
asv_abund_matrix,
retrieve_files=FALSE,
overwrite_existing = TRUE
)
#> Tracking read counts:
#> samplename_barcode input filtered denoisedF denoisedR merged nonchim
#> 1 S1_its 299 163 146 141 132 132
#> 2 S2_its 235 144 113 99 99 99
#> Tracking read counts:
#> samplename_barcode input filtered denoisedF denoisedR merged nonchim
#> 1 S1_rps10 196 145 145 145 145 145
#> 2 S2_rps10 253 182 181 181 181 181
# }