Make an amplified sequence variant (ASV) abundance matrix for each of the input barcodes
Source:R/make_asv_abund_matrix.R
make_asv_abund_matrix.RdMake an amplified sequence variant (ASV) abundance matrix for each of the input barcodes
Details
The function processes data for each unique barcode separately, inferring ASVs, merging reads, and creating an ASV abundance matrix. To do this, the 'DADA2' core denoising alogrithm is used to infer ASVs.
Examples
# \donttest{
# The primary wrapper function for 'DADA2' ASV inference steps
analysis_setup <- prepare_reads(
data_directory = system.file("extdata", package = "demulticoder"),
output_directory = tempdir(),
overwrite_existing = TRUE
)
#> Existing files found in the output directory. Overwriting existing files.
#> Rows: 2 Columns: 25
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: ","
#> chr (3): primer_name, forward, reverse
#> dbl (18): minCutadaptlength, maxN, maxEE_forward, maxEE_reverse, truncLen_fo...
#> lgl (4): already_trimmed, count_all_samples, multithread, verbose
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Rows: 2 Columns: 25
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: ","
#> chr (3): primer_name, forward, reverse
#> dbl (18): minCutadaptlength, maxN, maxEE_forward, maxEE_reverse, truncLen_fo...
#> lgl (4): already_trimmed, count_all_samples, multithread, verbose
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Rows: 4 Columns: 3
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: ","
#> chr (3): sample_name, primer_name, organism
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Creating output directory: /tmp/RtmpAtZc28/demulticoder_run/prefiltered_sequences
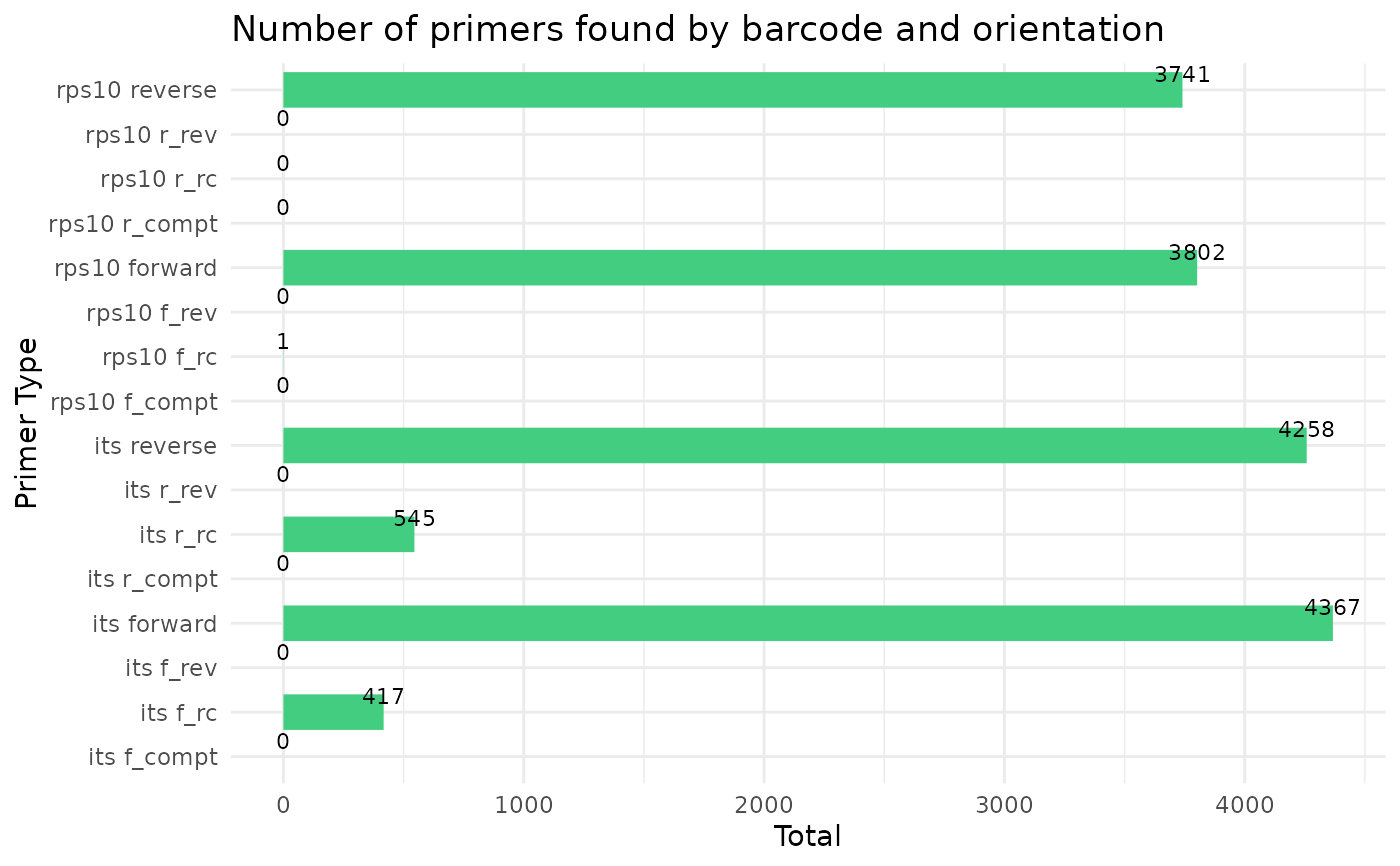 cut_trim(
analysis_setup,
cutadapt_path="/usr/bin/cutadapt",
overwrite_existing = TRUE
)
#> Running cutadapt 3.5 for its sequence data
cut_trim(
analysis_setup,
cutadapt_path="/usr/bin/cutadapt",
overwrite_existing = TRUE
)
#> Running cutadapt 3.5 for its sequence data
 #> Running cutadapt 3.5 for rps10 sequence data
#> Running cutadapt 3.5 for rps10 sequence data
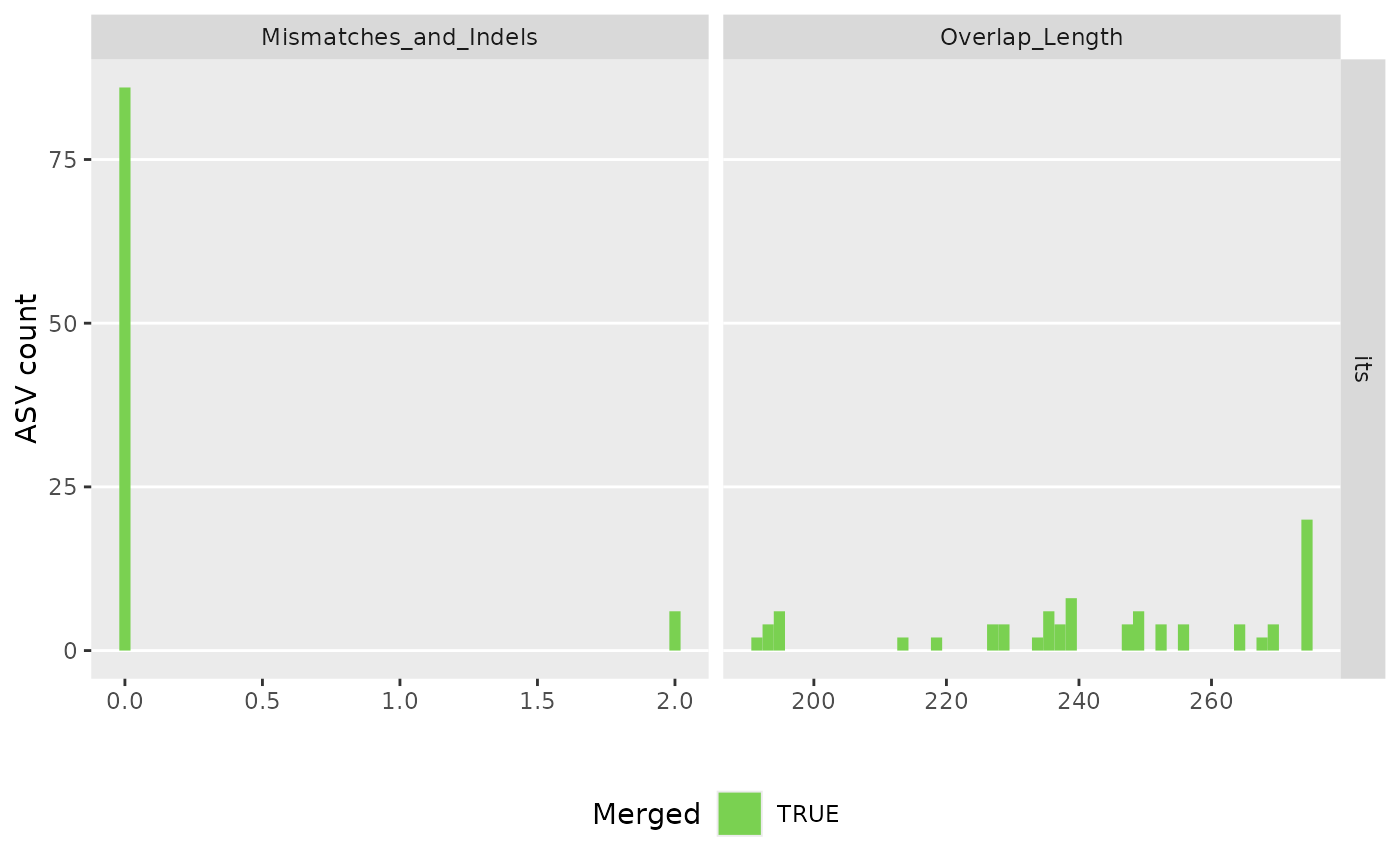 make_asv_abund_matrix(
analysis_setup,
overwrite_existing = TRUE
)
#> 80608 total bases in 307 reads from 2 samples will be used for learning the error rates.
#> Error rate plot for the Forward read of primer pair its
#> Warning: log-10 transformation introduced infinite values.
#> Sample 1 - 163 reads in 84 unique sequences.
#> Sample 2 - 144 reads in 96 unique sequences.
#> 82114 total bases in 307 reads from 2 samples will be used for learning the error rates.
#> Error rate plot for the Reverse read of primer pair its
#> Warning: log-10 transformation introduced infinite values.
#> Sample 1 - 163 reads in 128 unique sequences.
#> Sample 2 - 144 reads in 119 unique sequences.
make_asv_abund_matrix(
analysis_setup,
overwrite_existing = TRUE
)
#> 80608 total bases in 307 reads from 2 samples will be used for learning the error rates.
#> Error rate plot for the Forward read of primer pair its
#> Warning: log-10 transformation introduced infinite values.
#> Sample 1 - 163 reads in 84 unique sequences.
#> Sample 2 - 144 reads in 96 unique sequences.
#> 82114 total bases in 307 reads from 2 samples will be used for learning the error rates.
#> Error rate plot for the Reverse read of primer pair its
#> Warning: log-10 transformation introduced infinite values.
#> Sample 1 - 163 reads in 128 unique sequences.
#> Sample 2 - 144 reads in 119 unique sequences.
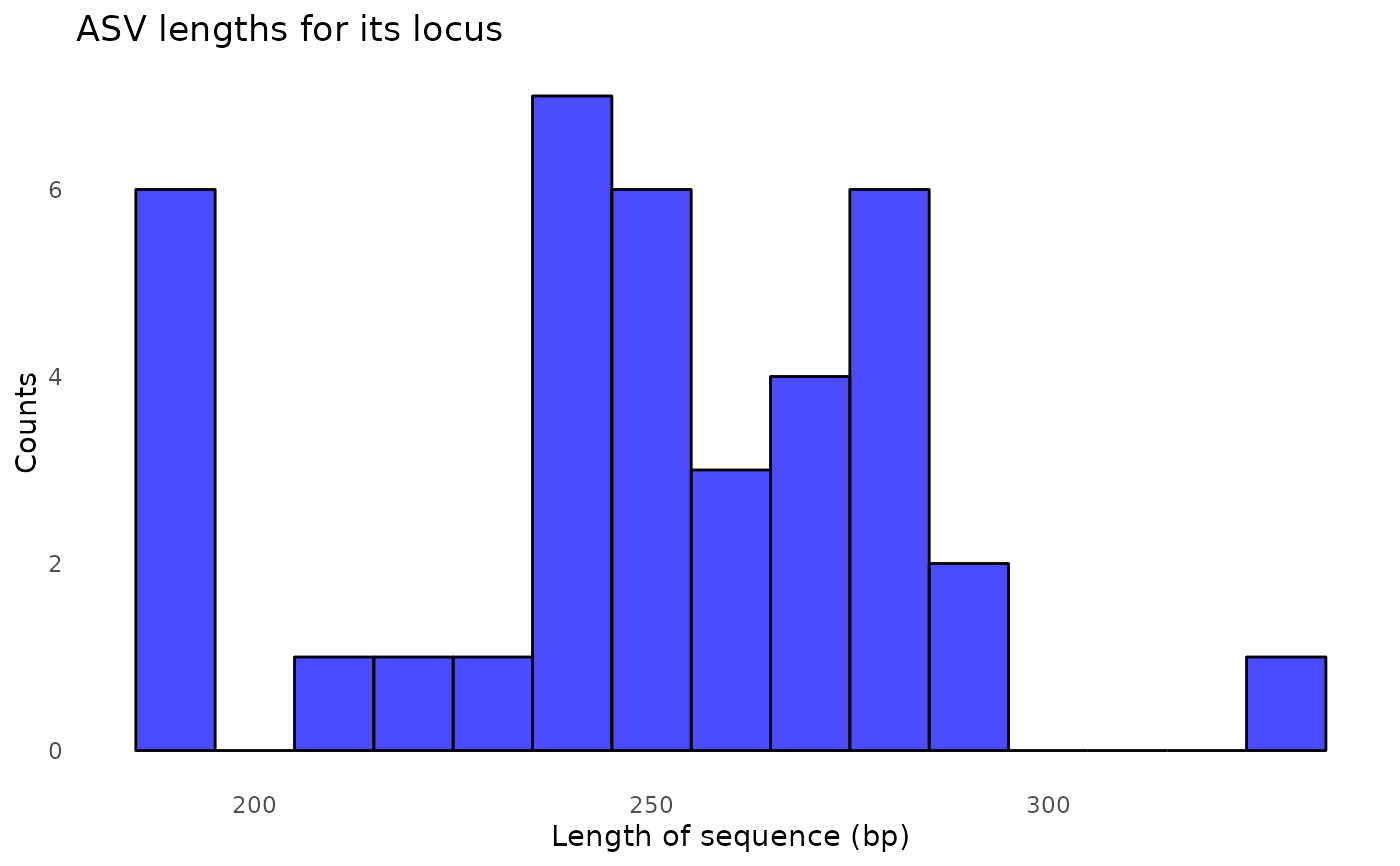
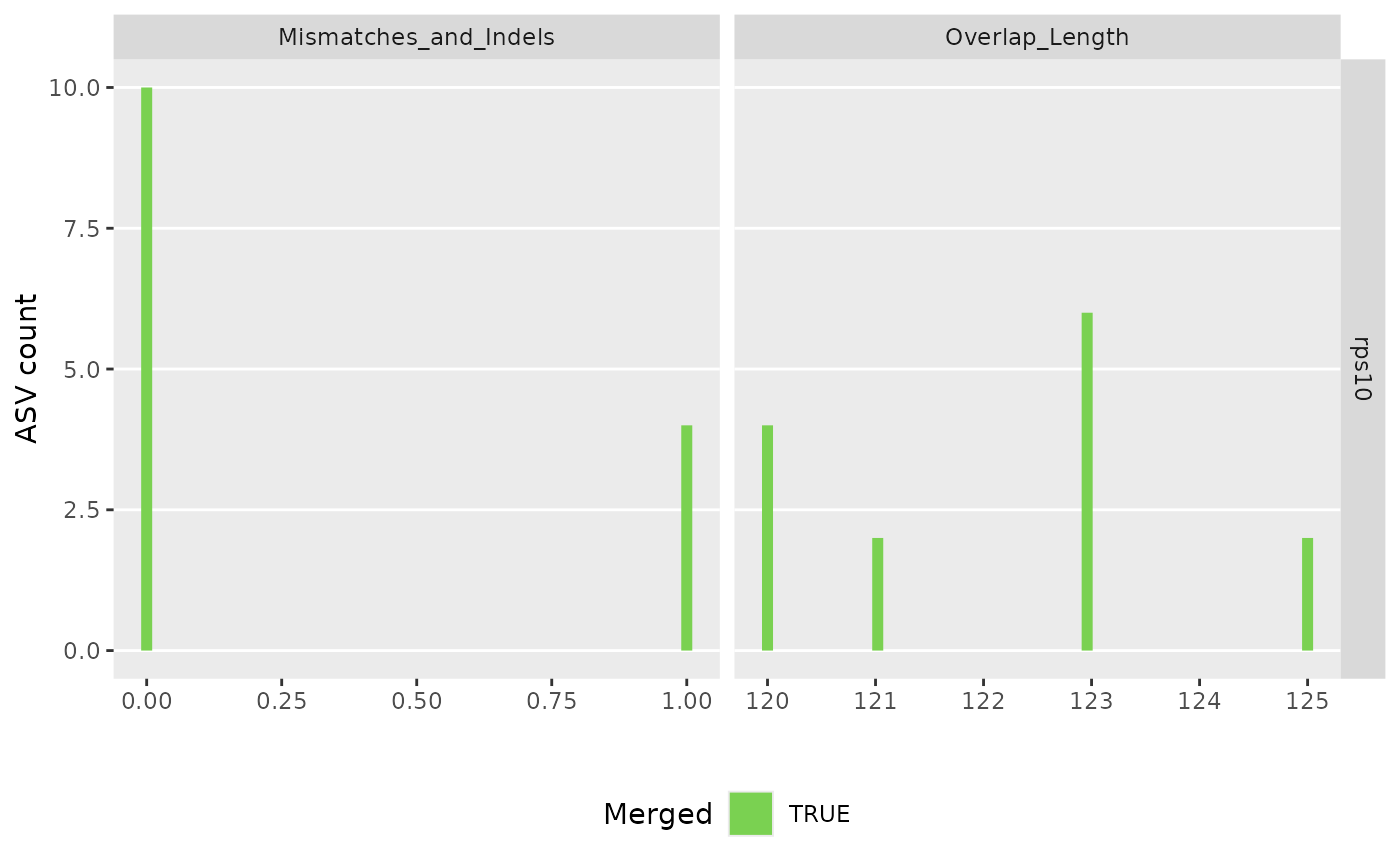 #> 91897 total bases in 327 reads from 2 samples will be used for learning the error rates.
#> Error rate plot for the Forward read of primer pair rps10
#> Warning: log-10 transformation introduced infinite values.
#> Sample 1 - 145 reads in 107 unique sequences.
#> Sample 2 - 182 reads in 133 unique sequences.
#> 91567 total bases in 327 reads from 2 samples will be used for learning the error rates.
#> Error rate plot for the Reverse read of primer pair rps10
#> Warning: log-10 transformation introduced infinite values.
#> Sample 1 - 145 reads in 114 unique sequences.
#> Sample 2 - 182 reads in 170 unique sequences.
#> 91897 total bases in 327 reads from 2 samples will be used for learning the error rates.
#> Error rate plot for the Forward read of primer pair rps10
#> Warning: log-10 transformation introduced infinite values.
#> Sample 1 - 145 reads in 107 unique sequences.
#> Sample 2 - 182 reads in 133 unique sequences.
#> 91567 total bases in 327 reads from 2 samples will be used for learning the error rates.
#> Error rate plot for the Reverse read of primer pair rps10
#> Warning: log-10 transformation introduced infinite values.
#> Sample 1 - 145 reads in 114 unique sequences.
#> Sample 2 - 182 reads in 170 unique sequences.
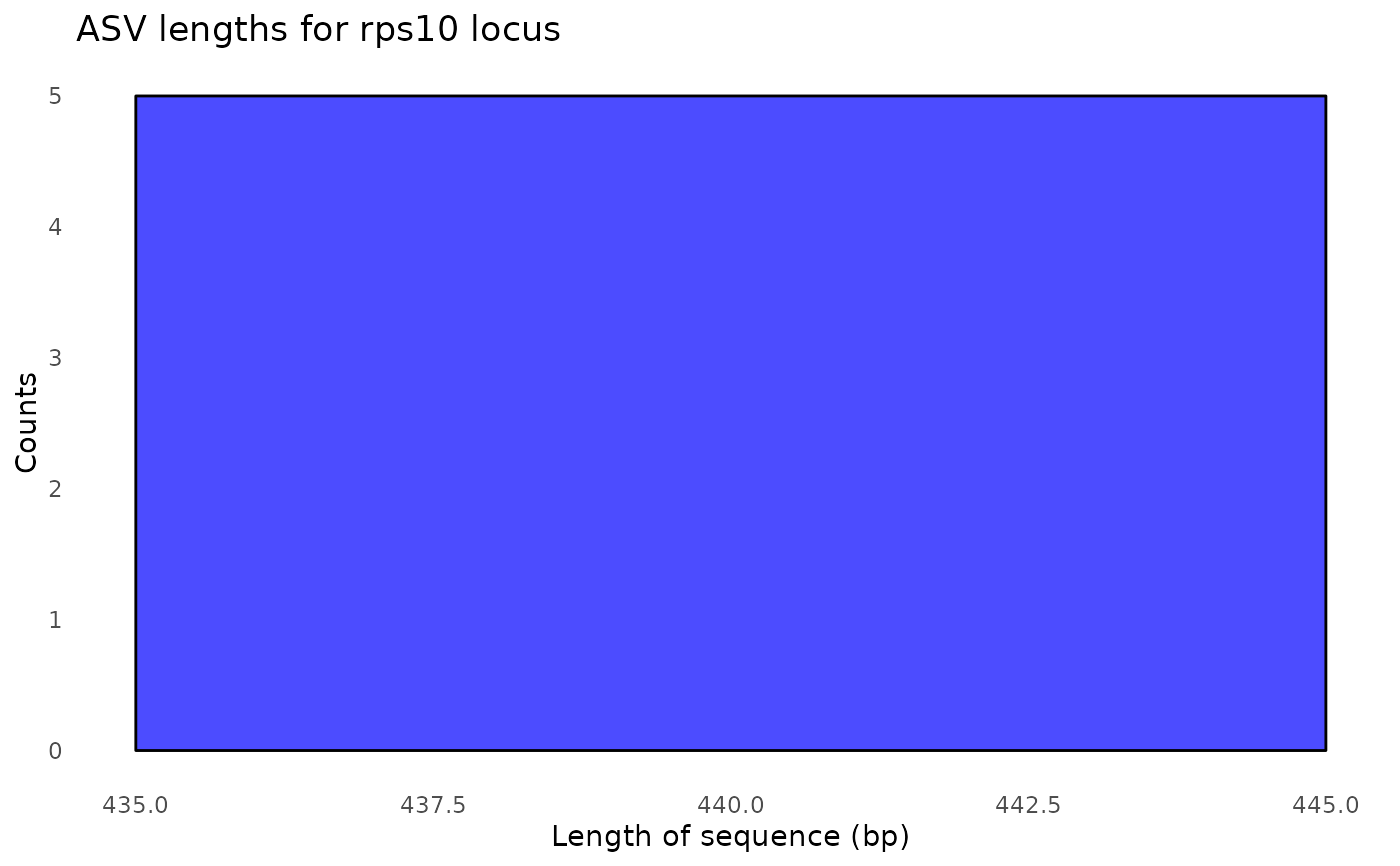
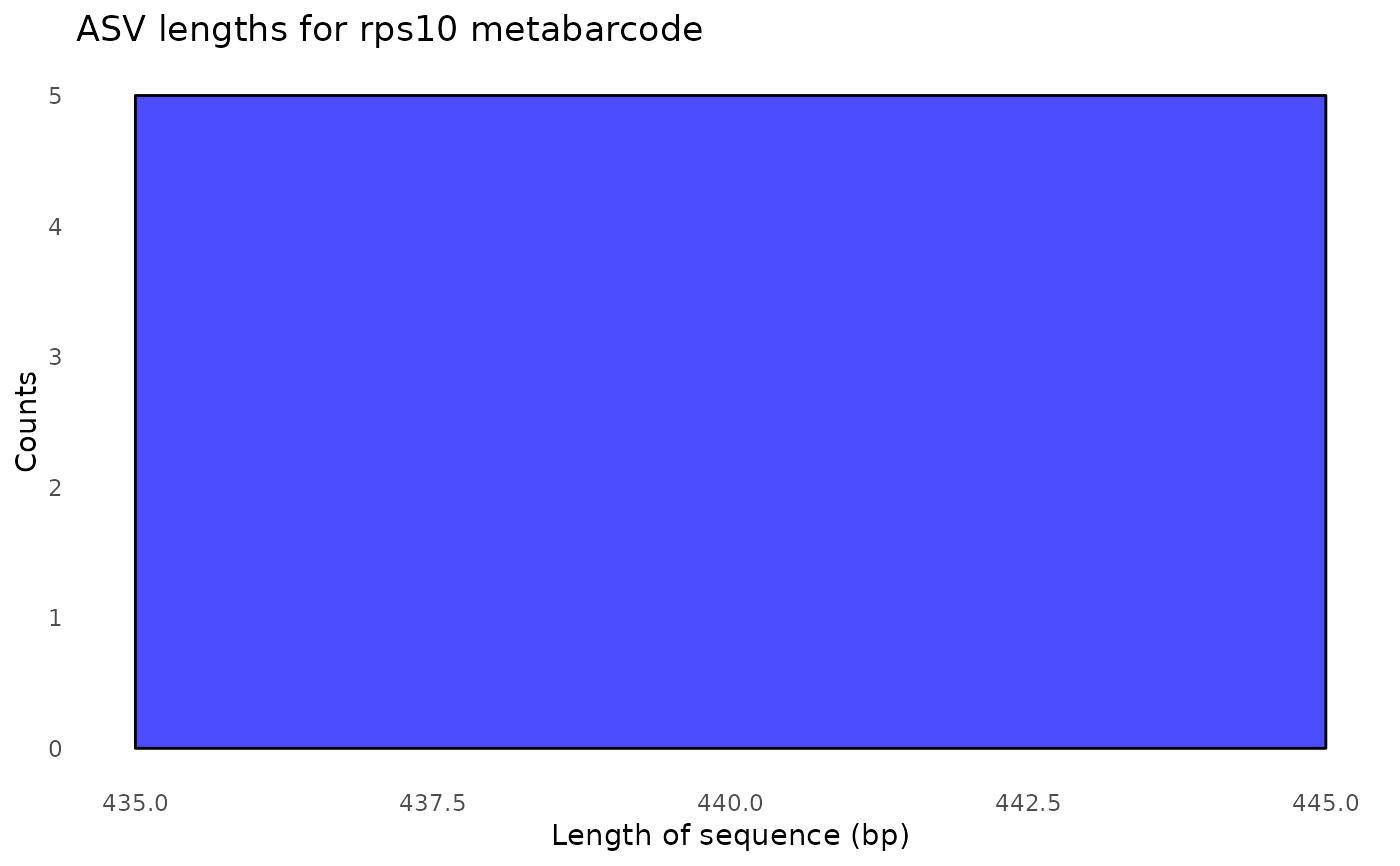 #> $its
#> [1] "/tmp/RtmpAtZc28/demulticoder_run/asvabund_matrixDADA2_its.RData"
#>
#> $rps10
#> [1] "/tmp/RtmpAtZc28/demulticoder_run/asvabund_matrixDADA2_rps10.RData"
#>
# }
#> $its
#> [1] "/tmp/RtmpAtZc28/demulticoder_run/asvabund_matrixDADA2_its.RData"
#>
#> $rps10
#> [1] "/tmp/RtmpAtZc28/demulticoder_run/asvabund_matrixDADA2_rps10.RData"
#>
# }