Main command to trim primers using 'Cutadapt' and core 'DADA2' functions
Source:R/cut_primers_trim_reads.R
cut_trim.RdMain command to trim primers using 'Cutadapt' and core 'DADA2' functions
Details
If samples are comprised of two different metabarcodes (like ITS1 and rps10), reads will also be demultiplexed prior to 'DADA2'-specific read trimming steps.
Examples
# \donttest{
# Remove remaining primers from raw reads, demultiplex pooled barcoded samples,
# and then trim reads based on specific 'DADA2' parameters
analysis_setup <- prepare_reads(
data_directory = system.file("extdata", package = "demulticoder"),
output_directory = tempdir(),
overwrite_existing = TRUE
)
#> Existing files found in the output directory. Overwriting existing files.
#> Rows: 2 Columns: 25
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: ","
#> chr (3): primer_name, forward, reverse
#> dbl (18): minCutadaptlength, maxN, maxEE_forward, maxEE_reverse, truncLen_fo...
#> lgl (4): already_trimmed, count_all_samples, multithread, verbose
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Rows: 2 Columns: 25
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: ","
#> chr (3): primer_name, forward, reverse
#> dbl (18): minCutadaptlength, maxN, maxEE_forward, maxEE_reverse, truncLen_fo...
#> lgl (4): already_trimmed, count_all_samples, multithread, verbose
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Rows: 4 Columns: 3
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: ","
#> chr (3): sample_name, primer_name, organism
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Creating output directory: /tmp/RtmpAtZc28/demulticoder_run/prefiltered_sequences
 cut_trim(
analysis_setup,
cutadapt_path="/usr/bin/cutadapt",
overwrite_existing = TRUE
)
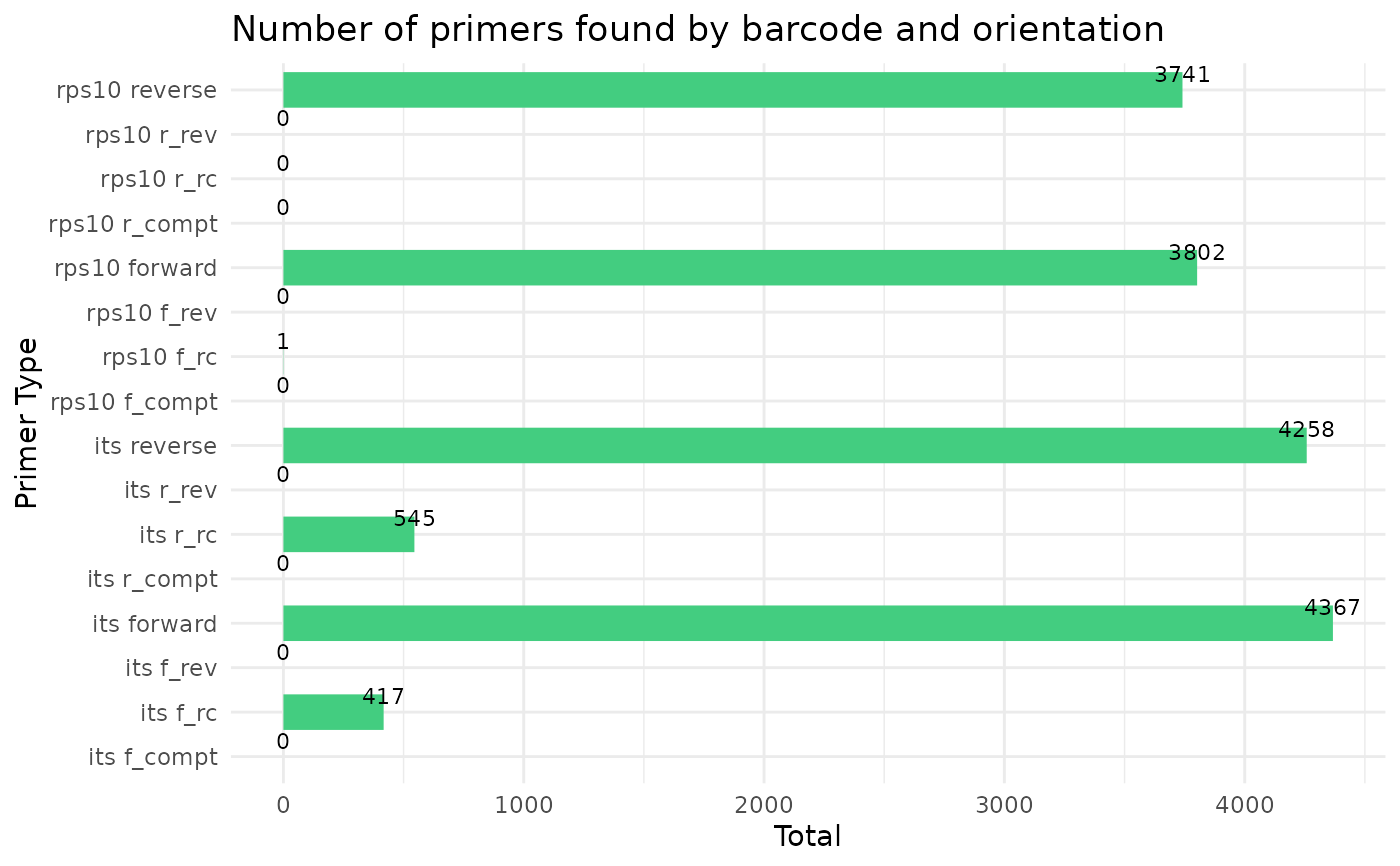
#> Running cutadapt 3.5 for its sequence data
cut_trim(
analysis_setup,
cutadapt_path="/usr/bin/cutadapt",
overwrite_existing = TRUE
)
#> Running cutadapt 3.5 for its sequence data
 #> Running cutadapt 3.5 for rps10 sequence data
#> Running cutadapt 3.5 for rps10 sequence data
 # }
# }