![]()
NOTE: THIS PROJECT IS UNDER DEVELOPMENT AND MAY NOT FUNCTION AS EXPECTED UNTIL THIS MESSAGE GOES AWAY
Introduction
nf-core/pathogensurveillance is a population genomic pipeline for pathogen diagnosis, variant detection, and biosurveillance. The pipeline accepts the paths to raw reads for one or more organisms (in the form of a CSV file) and creates reports in the form of interactive HTML reports or PDF documents. Significant features include the ability to analyze unidentified eukaryotic and prokaryotic samples, creation of reports for multiple user-defined groupings of samples, automated discovery and downloading of reference assemblies from NCBI RefSeq, and rapid initial identification based on k-mer sketches followed by a more robust core genome phylogeny and SNP-based phylogeny.
Usage
[!NOTE] If you are new to Nextflow and nf-core, please refer to this page on how to set-up Nextflow. Make sure to test your setup with
-profile testbefore running the workflow on actual data.
First, prepare a samplesheet with your input data that looks as follows:
nextflow run nf-core/pathogensurveillance -r dev -profile RUN_TOOL,xanthomonas_small -resume --out_dir test_outputNote that some form of configuration will be needed so that Nextflow knows how to fetch the required software. This is usually done in the form of a config profile (RUN_TOOL in the example command above). You can chain multiple config profiles in a comma-separated string.
- The pipeline comes with config profiles called
docker,singularity,podman,shifter,charliecloudandcondawhich instruct the pipeline to use the named tool for software management. For example,-profile xanthomonas_small,docker.- Please check nf-core/configs to see if a custom config file to run nf-core pipelines already exists for your Institute. If so, you can simply use
-profile <institute>in your command. This will enable eitherdockerorsingularityand set the appropriate execution settings for your local compute environment.- If you are using
singularity, please use thenf-core downloadcommand to download images first, before running the pipeline. Setting theNXF_SINGULARITY_CACHEDIRorsingularity.cacheDirNextflow options enables you to store and re-use the images from a central location for future pipeline runs.- If you are using
conda, it is highly recommended to use theNXF_CONDA_CACHEDIRorconda.cacheDirsettings to store the environments in a central location for future pipeline runs.
–>
Now, you can run the pipeline using:
nextflow run nf-core/pathogensurveillance -r dev -profile RUN_TOOL -resume --sample_data <TSV/CSV> --out_dir <OUTDIR> --download_bakta_dbnextflow run nf-core/pathogensurveillance \
-profile <docker/singularity/.../institute> \
--input samplesheet.tsv \
--outdir <OUTDIR>Background - why use pathogensurveillance?
TL;DR: unknown gDNA FASTQ -> sample ID + phylogeny + publication-quality figures
Most genomic tools are designed to be used with a reference genome. Yet this is at odds with the work of pathogen diagnosticians, who often deal with unknown samples. Finding the right reference manually may be cumbersome and require a suprising amount of technical skill.
PathogenSurveillance picks a good reference genome for you. It does this through the program sourmash. In simple terms, this takes a sample’s DNA “fingerprint” and finds the closest match in a “DNA fingerprint library” spanning the tree of life. In more technical terms, the pipeline generates k-mer sketches from your reads assembled into genomes, then uses the identified reference to do a boilerplate, but robust phylogenetic analysis of your submitted samples.
In our experience, the pipeline usually chooses the best possible reference genome. At a minimum it will choose a reference that is good enough to build an informative phylogeny and allow you to see the contextual placement your samples.
Pathogensurveillance is designed to use as many types of genomic DNA input as possible. It works for common shortread and longread sequencing technologies and for both prokaryotes and eukaryotes. There is a good deal of emergent complexity required to work with such a broad sample range, but pathogensurveillance handles this automatically.
While PathogenSurveillance may be a useful tool for researchers of all levels, it was designed with those who may have limited bioinformatics training in mind. Pathogensurveillance is very simple to run. At a minimum, all that needs to be supplied is a .CSV file with a single column specifying the path to your sample’s sequencing reads. Other information is optional, but if provided will used to customize the output report or conditionally use particular reference genomes.
pathogensurveillance is particularly good for:
- unknown sample identification
- exploratory population analysis using minimal input parameters
- inexperienced bioinformatics users
- efficient parallelization of tasks
- repeated analysis (given caching) where you would like to add new samples to a past run
pathogensurveillance works for non pathogens too!
pathogensurveillance is not designed for:
- viral sequence
- non gDNA datasets (DNA assembly fasta files, RNA-seq, RAD-seq, ChIP-seq, etc.)
- mixed/impure samples (this may change in future versions)
- Highly specialized population genetic analysis, or researchers who would like to extensively test parameters at each stage
Pipeline summary
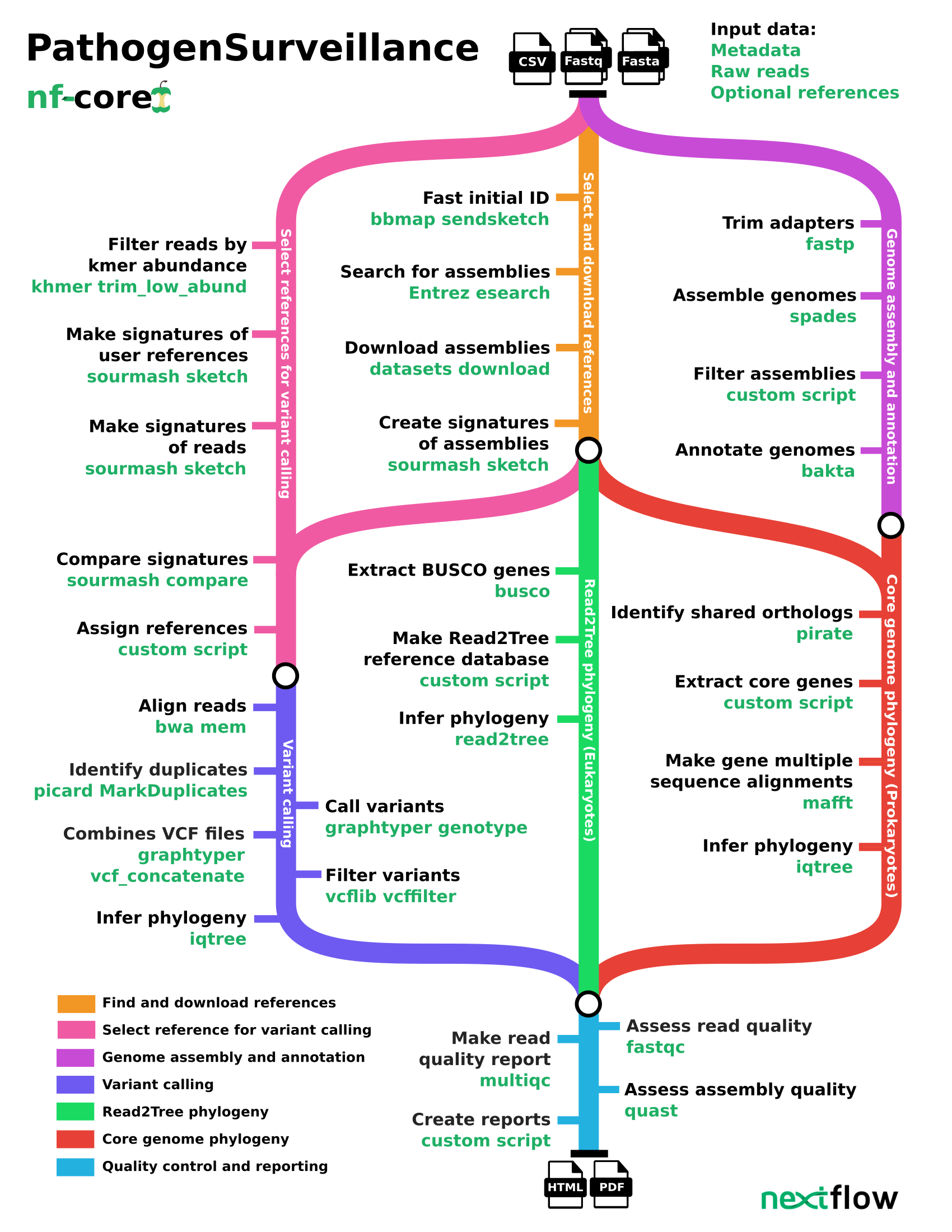
This is a quick breakdown of the processes used by pathogensurveillance:
- Download sequences and references if they are not provided locally
- Genome assembly
- Illumina shortreads: (
spades) - Pacbio or Oxford Nanopore longreads: (
flye)
- Illumina shortreads: (
- Quickly obtain several initial sample references (
bbmap) - More accurately select appropriate reference genomes. Genome “sketches” are compared between first-pass references, samples, and any references directly provided by the user (
sourmash) - Genome annotation (
bakta) - Align reads to reference sequences (
bwa) - Variant calling and filtering (
graphtyper,vcflib) - Determine relationship between samples and references
- Build SNP tree from variant calls (
iqtree) - For Prokaryotes:
- Identify shared orthologs (
pirate) - Build tree from core genome phylogeny (
iqtree)
- Identify shared orthologs (
- For Eukaryotes:
- Identify BUSCO genes (
busco) - Build tree from BUSCO genes (
iqtree)
- Identify BUSCO genes (
- Build SNP tree from variant calls (
- Generate interactive html report/pdf file
- Sequence and assembly information (
fastqc, multiqc, quast) - sample identification tables and heatmaps
- Phylogenetic trees from genome-wide SNPs and core genes
- minimum spanning network
- Sequence and assembly information (